

Welcome to UChicago PCCM Research!
Research defining the mechanisms, diagnosis, and treatment of lung disease and critical illness is a central mission of our group. This research is conducted in our clinical facilities as well as our basic research laboratories. Investigators are actively engaged in studying lung biology and cellular function related to critical illness, using techniques of cell and molecular biology, immunology, and genetics. Large and innovative clinical studies are also in place, with questions related to diagnosis and management of diverse diseases answered by accomplished clinical scientists observing individual patients and patient populations.
The strength of our research enterprise derives from the high degree of collaboration between basic and clinical scientists, and the joining of faculty and research associates with diverse training, expertise, and interests. The PhD, MD, and MD-PhD investigators in our group sustain this multi-faceted and dynamic approach to biomedical research. The essence of this activity is to ask important questions, and to answer them with whatever means are necessary—often employing the means of both clinical and basic science. The flow of ideas and findings is from bedside to bench and back, with each discovery adding to new questions requiring creative approaches to answer them. This exciting atmosphere provides abundant learning opportunities for students at every level of training, and we have successfully incorporated college, medical, and graduate students; post-doctoral scientists; residents; and fellows in our many programs.
News
Single cell metabolism methods paper is out in STAR protocols (Fang/Wu labs!)
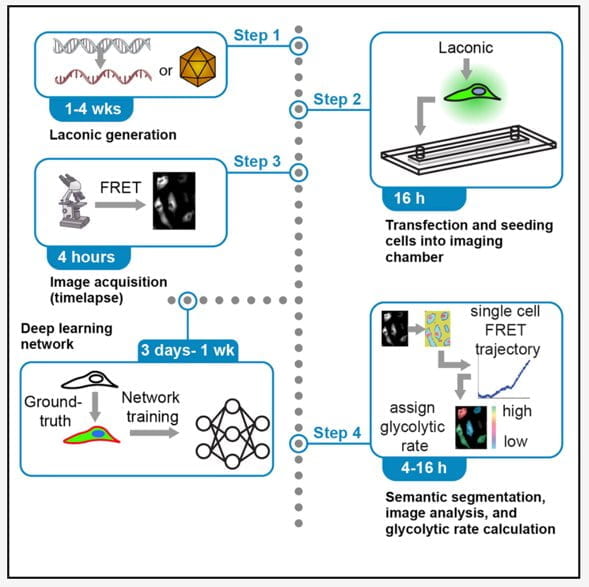
Single-cell lactate production rate as a measure of glycolysis in endothelial cells
Devin Harrison,1,2,3,4 David Wu,2,4,5 Jun Huang,1,3,* and Yun Fang1,2,6,**
1Graduate Program in Biophysical Sciences, The University of Chicago, Chicago, IL 60637, USA
2Department of Medicine, Biological Sciences Division, The University of Chicago, Chicago, IL 60637, USA
3Pritzker School of Molecular Engineering, The University of Chicago, Chicago, IL 60637, USA
4These authors contributed equally
5Technical contact
6Lead contact
*Correspondence: huangjun@uchicago.edu
**Correspondence: yfang1@medicine.bsd.uchicago.edu
Summary
Heterogeneous metabolism supports critical single-cell functions. Here, we describe deep-learning-enabled image analyses of a genetically encoded lactate-sensing probe which can accurately quantify metabolite levels and glycolytic rates at the single-cell level. Multiple strategies and test data have been included to obviate possible obstacles including successful sensor expression and accurate segmentation. This protocol reliably discriminates between metabolically diverse subpopulations which can be used to directly link metabolism to functional phenotypes by integrating spatiotemporal information, genetic or pharmacological perturbations, and real-time metabolic states.
New publication by the Mutlu lab!
Congratulations to Regul and company!
Rengul Cetin-Atalay, Angelo Y. Meliton, David Wu, Lucas M. Kimmig, Parker S. Woods, Robert B. Hamanaka, Ying-Jie Peng, Jayasri Nanduri, Nanduri Prabhakar, and Gökhan M. Mutlu. Intermittent hypoxia-induced activation of endothelial cells is mediated via sympathetic activation dependent catecholamine release. Accepted in Frontiers in Physiology. 2021.
Abstract:
Obstructive sleep apnea (OSA) is a common breathing disorder affecting a significant percentage of the adult population. OSA is an independent risk factor for cardiovascular disease (CVD); however, the underlying mechanisms are not completely understood. Since the severity of hypoxia correlates with some of the cardiovascular effects, intermittent hypoxia (IH) is thought to be one of the mechanisms by which OSA may cause CVD. Here, we investigated the effect of IH on endothelial cell (EC) activation, characterized by the expression of inflammatory genes, that is known to play an important role in the pathogenesis of CVD. Exposure of C57BL/6 mice to IH led to aortic EC activation, while in vitro exposure of ECs to IH failed to do so, suggesting that IH does not induce EC activation directly, but indirectly. One of the consequences of IH is activation of the sympathetic nervous system and catecholamine release. We found that exposure of mice to IH caused elevation of circulating levels of catecholamines. Inhibition of the IH-induced increase in catecholamines by pharmacologic inhibition or by adrenalectomy or carotid body ablation prevented the IH-induced EC activation in mice. Supporting a key role for catecholamines, epinephrine alone was sufficient to cause EC activation in vivo and in vitro. Together, these results suggested that IH does not directly induce EC activation, but does so indirectly via release of catecholamines. These results suggest that targeting IH-induced sympathetic nerve activity and catecholamine release may be a potential therapeutic target to attenuate the CV effects of OSA.